Mit Künstlicher Intelligenz die „Fingerabdrücke“ von Molekülen errechnen
HZB-Team nutzt selbstlernende Graphische Neuronale Netze, um experimentell gewonnene Messdaten korrekt zu interpretieren
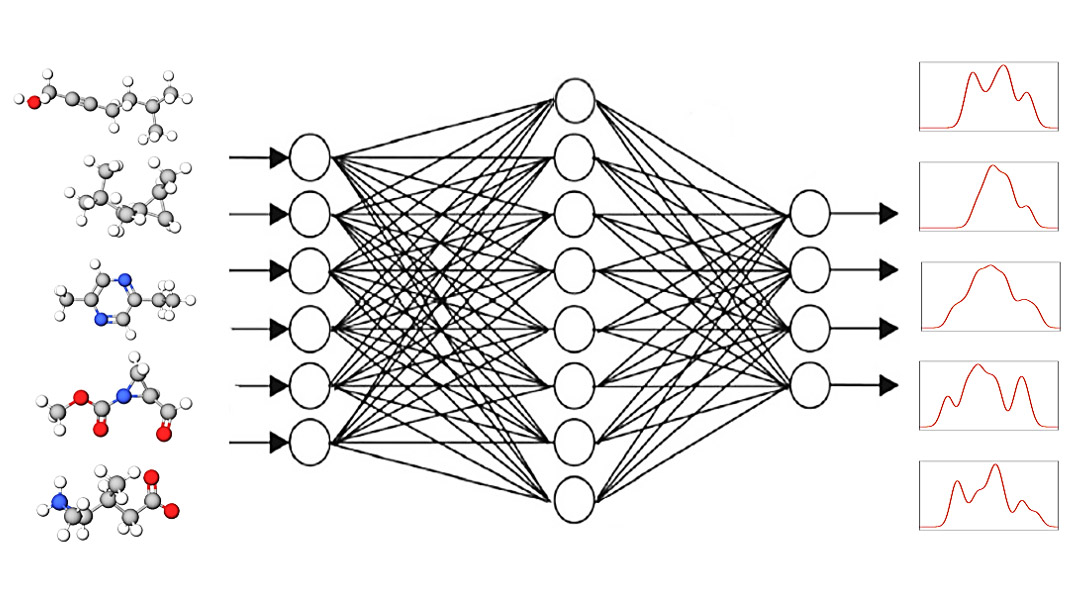
Mit konventionellen Methoden ist es extrem aufwändig, den spektralen Fingerabdruck von größeren Molekülen zu berechnen. Dies ist aber eine Voraussetzung, um experimentell gewonnene Messdaten korrekt zu interpretieren. Nun hat ein Team am HZB mit selbstlernenden Graphischen Neuronalen Netzen sehr gute Ergebnisse in deutlich kürzerer Zeit erzielt.
„Biomoleküle, große anorganische Moleküle, aber auch Quantenpunkte, die oft aus tausenden von Atomen bestehen, sind mit konventionellen Methoden wie der DFT kaum noch vorab zu berechnen“, sagt PD Dr. Annika Bande, theoretische Chemikerin am HZB. Mit ihrem Team hat sie nun systematisch untersucht, wie sich die Rechenzeit durch den Einsatz von Methoden aus der Künstlichen Intelligenz verkürzen lässt.
Die Idee: Ein Computerprogramm aus der Gruppe der „graphischen neuronalen Netze“ oder GNN erhält als Input kleine Moleküle mit der Aufgabe, deren spektrale Antworten zu ermitteln. Im nächsten Schritt vergleicht das GNN-Programm die errechneten Spektren mit den bekannten Zielspektren (DFT oder experimentell) und korrigiert in der folgenden Runde den Berechnungsweg entsprechend. Runde für Runde wird so das Ergebnis immer besser. Das GNN-Programm lernt also selbstständig mit Hilfe bekannter Spektren, wie sich Spektren zuverlässig berechnen lassen.
„Wir haben fünf neuere GNN trainiert und festgestellt, dass sich mit einem davon, dem SchNet-Modell, enorme Verbesserungen erreichen lassen: Die Genauigkeit steigt um 20 % und dies in einem Bruchteil der Rechenzeit“, sagt Erstautor Kanishka Singh. Singh nimmt an der Graduiertenschule HEIBRiDS teil und wird in diesem Rahmen sowohl von Informatik-Experten Prof. Ulf Leser aus der Humboldt-Universität zu Berlin als auch von Annika Bande betreut.
„Kürzlich entwickelte GNN-Frameworks könnten sogar noch besser abschneiden“, meint die theoretische Chemikerin. „Und die Nachfrage ist sehr groß. Wir wollen diese Forschungsrichtung daher vertiefen und planen dafür ab Sommer eine neue Postdoc-Stelle im Rahmen des Helmholtz-Projekts eXplainable Artificial Intelligence for X-ray Absorption Spectroscopy ein.“
Publikation:
Graph Neural Networks for Learning Molecular Excitation Spectra
Kanishka Singh, Jannes Münchmeyer, Leon Weber, Ulf Leser and Annika Bande
Journal of Chemical Theory and Computation (2022). DOI: 10.1021/acs.jctc.2c00255
Anmerkung:
Die Arbeit entstand im Rahmen der Graduiertenschule HEIBRiDS und wird im Helmholtz-Projekt „eXplainable Artificial Intelligence for X-ray Absorption Spectroscopy“ (XAI-4-XAS) weitergeführt. Im Kern geht es in diesem Helmholtz-Projekt (mit HEREON, Leitung HZB) darum, die GNN auch auf sehr große Moleküle auszudehnen. Dies soll in Kombination mit der probabilistischen Analyse von Molekülmotiven erreicht werden. Sie dient dazu, nur den Teil des Konfigurationsphasenraums der Moleküle zu erfassen, der für die genaue Vorhersage von Röntgenspektren erforderlich ist. Die Ergebnisse der Vorhersagen ermöglichen eine rigorose Interpretation von XAS-Experimenten.
Kontakt:
Helmholtz-Zentrum Berlin für Materialien und Energie
Nachwuchsgruppe Theorie der Elektronendynamik und Spektroskopie
Dr. Annika Bande
Tel.: +49 30 8062-42026
annika.bande(at)helmholtz-berlin.de
Pressestelle:
Dr. Antonia Rötger
Tel.: +49 30 8062-43733
E-Mail antonia.roetger(at)helmholtz-berlin.de
Pressemitteilung HZB vom 13.06.2022